NanoLaf (Use of mRNA-loaded polymeric nanoparticles as an innovative replacement therapy to treat Lafora Disease)
Polymeric nanoparticles to deliver mRNA as replacement therapy in Lafora Disease
Other entities
Individual project
01/01/2023 a 31/12/2024
126.400€
ACTIVE
Genetic and rare diseases (GARDs) represent a large category of disorders which share the similarity of each affecting a small portion of the population but altogether affecting more than 350 million patients worldwide and remaining a significant challenge in the clinic. While a single rare disease may affect only a small group of people, the collective impact of rare diseases can be enormous.
Recent decades have witnessed the impressive progress in the fight against GARDs, with an improved understanding of the genetic origins of rare diseases and the rapid development in gene therapy providing a new avenue for GARD therapy. RNA-based therapeutics, such as RNA interference (RNAi), messenger RNA (mRNA) and RNA-involved genome editing technologies, demonstrate great potential as a therapy tool for treating genetic associated rare diseases.
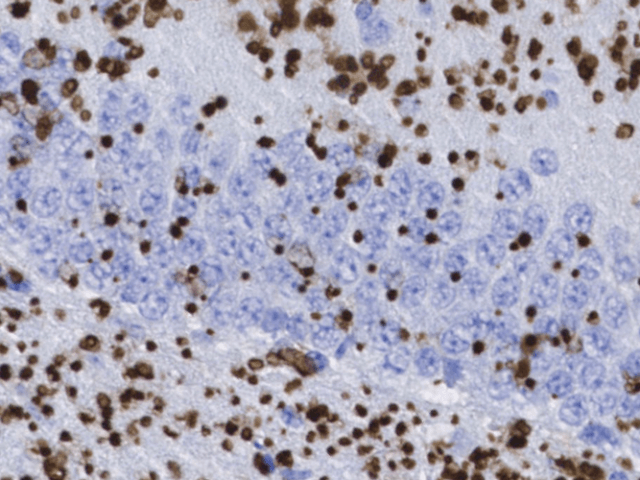
Lafora disease (LD, OMIM#254780, ORPHA#501) is a devastatingly fatal, autosomal recessive form of childhood dementia affecting similar rates of men and women. The onset of the disease is in adolescence, in apparently healthy teenagers, with headaches, seizures, and insidious decline in cognitive function. No effective treatment is known, and the disease progresses rapidly with amplification of seizures, loss of neurologic functions and dementia, inevitably leading to the death of the patient 5-10 years after the onset.
The hallmark of the disease is the accumulation of cytoplasmic glycogen deposits, known as Lafora bodies (LBs), in the brain and other tissues. The disease is caused by mutations in one of two genes: EPM2A, which encodes the glycogen phosphatase laforin, and EPM2B, which encodes the E3-ubiquitin ligase malin.
LD, which is characterized by the accumulation of cytoplasmic glycogen deposits, known as Lafora bodies (LBs), in the brain and other tissues. We have demonstrated that the accumulation of glycogen in the form of LBs is the cause and driver of LD pathophysiology, including: seizures, neuronal loss, inflammation, aberrant brain metabolism, and behavioral abnormalities.
We have described a new generation of polymers (poly-(beta amino-esters), pBAEs), ideal for gene delivery thanks to their efficiency in encapsulating and protecting mRNA and selectively delivering it to target cells, where the codified protein is efficiently expressed
Our main objectives are:
1. Assess the efficacy of laforin re-introduction as a gene therapy approach for laforin-deficient LD.
2. Develop a new-generation of actively targeted ZWO-pBAE nanoparticles loaded with malin/laforin mRNA as a preclinical proof-of-concept replacement therapy
PRIMARY INVESTIGATOR

LINKED NEWS