Chelsea’s Hope (Neurodegeneracion Lafora)
Role específico de astrocitos y neuronase en la enfermedad de Lafora.
Other entities

Individual project
24/08/2021 a 24/08/2022
11.000€
COMPLETED

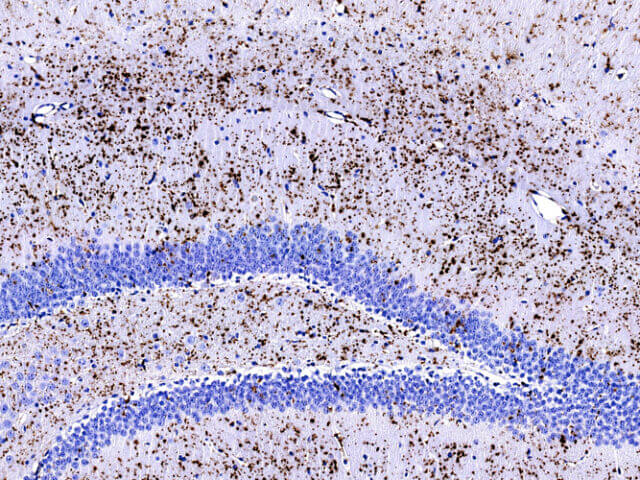
La enfermedad de Lafora es una patología rara que, pese a su baja prevalencia, supone graves consecuencias para los pacientes y sus cuidadores. Así pues, resulta necesario continuar la investigación para clarificar los mecanismos subyacentes y desarrollar nuevos tratamientos paliativos y curativos de la enfermedad.
La enfermedad de Lafora es una forma de epilepsia mioclónica progresiva de herencia autosómica recesiva, de inicio en la infancia tardía o en la adolescencia, y producida por mutaciones de pérdida de función en los genes EPM2A o EPM2B, los cuales codifican para las proteínas laforina y malina, respectivamente.
Los principales síntomas de la enfermedad, que empeoran progresivamente, son mioclonías, crisis occipitales, crisis tonicoclónicas generalizadas, deterioro cognitivo, síntomas neuropsiquiátricos y ataxia. El curso es progresivo y fatal. Patológicamente, se caracteriza por la presencia de depósitos de poliglucosanos (denominados cuerpos de Lafora) en el cerebro, el hígado, el músculo y las glándulas sudoríparas. El diagnóstico de enfermedad de Lafora se realiza mediante hallazgos clínicos, electrofisiológicos, histológicos y genéticos. En la actualidad no existe un tratamiento que erradique o prevenga su desarrollo. Tradicionalmente, se utilizan fármacos antiepilépticos para el tratamiento de las mioclonías y las convulsiones, aunque aparecen resistencias a éstas.
PRIMARY INVESTIGATOR

LINKED NEWS